
Expert Insight for Understanding Scoliosis in Children
Scoliosis is a common condition of the spine, often found in children and young adolescents. Questions about the condition are among the most frequently asked…

Update your location to show providers, locations, and services closest to you.
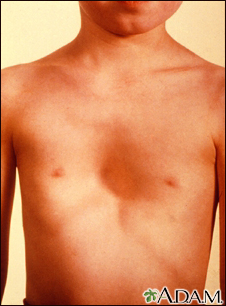
Marfan syndrome is a disorder of connective tissue. This is the tissue that strengthens the body's structures.
Disorders of connective tissue affect the skeletal system, cardiovascular system, eyes, and skin.
Aortic aneurysm - Marfan
Marfan syndrome is caused by defects in a gene called fibrillin-1. Fibrillin-1 plays an important role as the building block for connective tissue in the body.
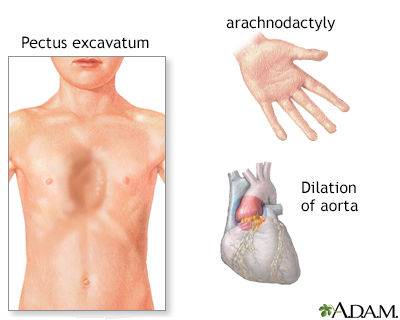
The gene defect also causes the long bones of the body to grow too much. People with this syndrome have tall height and long arms and legs. How this overgrowth happens is not well understood.
Other areas of the body that are affected include:
In most cases, Marfan syndrome is passed down through families (inherited). However, up to 30% of people have no family history, which is called "sporadic." In sporadic cases, the syndrome is believed to be caused by a new gene change.
People with Marfan syndrome are often tall with long, thin arms and legs and spider-like fingers (called arachnodactyly). The length of the arms is greater than height when arms are stretched out.
Other symptoms include:
Many people with Marfan syndrome suffer from chronic muscle and joint pain.
The health care provider will perform a physical exam. The joints may move around more than normal. There may also be signs of:
An eye exam may show:
The following tests may be performed:
An echocardiogram or another test should be done every year to look at the base of the aorta and possibly the heart valves. Depending on the results, you may need this test less often than yearly.
Vision problems should be treated when possible.
Monitor for scoliosis, especially during the teenage years.
Medicine to slow the heart rate and lower blood pressure may help prevent stress on the aorta. To avoid injuring the aorta, people with the condition may have to modify their activities. Some people may need surgery to replace the aortic root and valve.
Pregnant women with Marfan syndrome must be monitored very closely because of the increased stress on the heart and aorta.
The Marfan Foundation -- marfan.org
Heart-related complications may shorten the lifespan of people with this disease. However, many people live into their 60s and beyond. Good care and surgery may further extend lifespan.
Complications may include:
Couples who have this condition and are planning to have children may want to talk to a genetic counselor before starting a family.
Spontaneous new gene mutations leading to Marfan (less than one third of cases) cannot be prevented. If you have Marfan syndrome, see your provider at least once every year.
Doyle JJ, Doyle AJ, Dietz HC. Marfan syndrome. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 722.
Madan-Khetarpal S, Arnold G, Ortiz D. Genetic disorders and dysmorphic conditions. In: Zitelli BJ, McIntire SC, Nowalk AJ, eds. Zitelli and Davis' Atlas of Pediatric Diagnosis. 8th ed. Philadelphia, PA: Elsevier; 2021:chap 1.
Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 26th ed. Philadelphia, PA: Elsevier; 2020:chap 244.
Scoliosis is a common condition of the spine, often found in children and young adolescents. Questions about the condition are among the most frequently asked…
A recent retrospective study published by a team at University of Florida Health has shown that the Florida Sleeve procedure — originally developed at UF…
