Definition
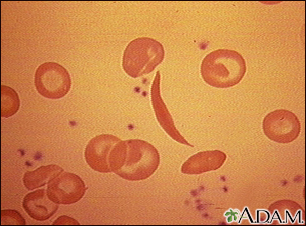
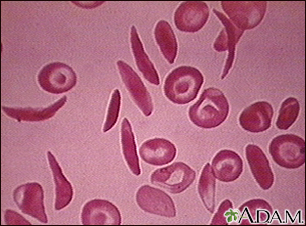
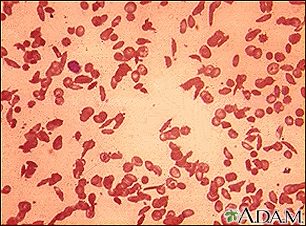
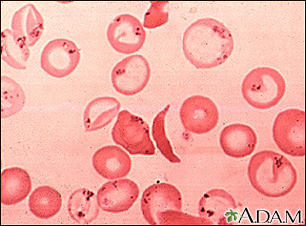
Sickle cell disease is a disorder passed down through families. The red blood cells that are normally shaped like a disk take on a sickle or crescent shape. Red blood cells carry oxygen throughout the body.
Alternative Names
Anemia - sickle cell; Hemoglobin SS disease (Hb SS); Sickle cell anemia
Causes
Sickle cell disease is caused by an abnormal type of hemoglobin called hemoglobin S. Hemoglobin is a protein inside red blood cells that carries oxygen.
- Hemoglobin S changes the red blood cells. The red blood cells become fragile and shaped like crescents or sickles.
- The abnormal cells deliver less oxygen to the body's tissues.
- They can also easily get stuck in small blood vessels and break into pieces. This can interrupt healthy blood flow and cut down even more on the amount of oxygen flowing to body tissues.
Sickle cell disease is inherited from both parents. If you get the sickle cell gene from only one parent, you will have sickle cell trait. People with sickle cell trait do not have the symptoms of sickle cell disease.
Sickle cell disease is much more common in people of African and Mediterranean descent. It is also seen in people from South and Central America, the Caribbean, and the Middle East.
Symptoms
Symptoms usually do not occur until after the age of 4 months.
Almost all people with sickle cell disease have painful episodes called crises. These can last from hours to days. Crises can cause pain in the lower back, leg, joints, and chest.
Some people have one episode every few years. Others have many episodes each year. The crises can be severe enough to require a hospital stay.
When sickle cell disease becomes more severe, symptoms may include:
- Fatigue
- Paleness
- Rapid heart rate
- Shortness of breath
- Yellowing of the eyes and skin (jaundice)
Younger children with sickle cell disease have attacks of abdominal pain.
The following symptoms may occur because small blood vessels become blocked by the abnormal cells:
- Painful and prolonged erection (priapism)
- Poor eyesight or blindness
- Problems with thinking or confusion caused by small strokes
- Ulcers on the lower legs (in adolescents and adults)
Over time, the spleen stops working. As a result, people with sickle cell disease may have symptoms of infections such as:
- Bone infection (osteomyelitis)
- Gallbladder infection (cholecystitis)
- Lung infection (pneumonia)
- Urinary tract infection
Other signs and symptoms include:
- Delayed growth and puberty
- Painful joints caused by arthritis
- Heart or liver failure due to too much iron (from blood transfusions)
Exams and Tests
Tests commonly done to diagnose and monitor people with sickle cell disease include:
Treatment
The goal of treatment is to manage and control symptoms, and to limit the number of crises. People with sickle cell disease need ongoing treatment, even when not having a crisis.
People with this condition should take folic acid supplements. Folic acid helps make new red blood cells.
Treatment for a sickle cell crisis includes:
- Blood transfusions (may also be given regularly to prevent stroke)
- Pain medicines
- Plenty of fluids
Other treatments for sickle cell disease may include:
- Hydroxyurea (Hydrea), which helps reduce the number of pain episodes (including breathing problems and risk for stroke) in some people
- Antibiotics, which help prevent bacterial infections that are common in children with sickle cell disease
- Medicines that reduce the amount of iron in the body
- Newer therapies to reduce the frequency and severity of pain crises have been approved
Treatments that may be needed to manage complications of sickle cell disease include:
Bone marrow or stem cell transplants can cure sickle cell disease, but this treatment is not an option for most people. People with sickle cell disease often cannot find well-matched stem cell donors.
People with sickle cell disease should have the following vaccinations to lower the risk for infection:
- Haemophilus influenzae vaccine (Hib)
- Pneumococcal conjugate vaccine (PCV)
- Pneumococcal polysaccharide vaccine (PPV)
Support Groups
Joining a support group where members share common issues can relieve the stress of a chronic disease.
Outlook (Prognosis)
In the past, people with sickle cell disease often died between ages 20 and 40. Thanks to modern care, people now can live to the age of 50 and beyond.
Causes of death include organ failure and infection.
When to Contact a Medical Professional
Contact your health care provider if you have:
- Any symptoms of infection (fever, body aches, headache, fatigue)
- Pain crises
- Painful and long-term erection (in men)
References
Howard J. Sickle cell disease and other hemoglobinopathies. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 26th ed. Philadelphia, PA: Elsevier; 2020:chap 154.
Meier ER. Treatment options for sickle cell disease. Pediatr Clin North Am. 2018;65(3)427-443. PMID 29803275 pubmed.ncbi.nlm.nih.gov/29803275/.
National Heart Lung and Blood Institute website. Evidence-based management of sickle cell disease: expert panel report, 2014. www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease. Updated September 2014. Accessed June 27, 2022.
Saunthararajah Y, Vichinsky EP. Sickle cell disease: clinical features and management. In: Hoffman R, Benz EJ, Silberstein LE, et al, eds. Hematology: Basic Principles and Practice. 7th ed. Philadelphia, PA: Elsevier; 2018:chap 42.
Smith-Whitley K, Kwiatkowski JL. Hemoglobinopathies. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 489.