The UF Lysosomal Storage Disease Program is committed to offering comprehensive and personalized clinical care for patients suffering from Lysosomal Storage Diseases (LSDs).
LSDs are genetic disorders that are caused by specific enzyme deficiencies resulting in multi-systemic disease, presenting with neurological, renal, cardiovascular, gastrointestinal, musculoskeletal, ophthalmological, and respiratory problems. The LSD program provides diagnosis, management, treatment, and monitoring of several different treatments available for patients affected by LSD including enzyme replacement therapy, small molecule therapy, hematopoietic stem cell therapy, and others.
UF clinical LSD program unites outstanding physicians from multiple specialties into a cooperative team devoted to offering excellence in patient care. Coy Heldermon, MD, coordinates the multi-disciplinary group from primary specialists in their respective sub-specialties. S. Parrish Winesett is the primary coordinator for pediatric patients. Our team has experience and resources to treat a wide range of complications commonly seen in these inherited metabolic diseases.
Based on the UF tradition of integrating clinical and basic research, teaching, and patient care, the LSD program is also involved in several clinical studies including clinical trials with the most advanced therapeutic agents. The unique characteristics of the program are an integrated approach to patients suffering from multi-systemic LSDs following UF principles of the pursuit of excellence and innovation in patient care.
The UF Lysosomal Storage Disease Program is directed by Coy Heldermon, MD. Dr. Heldermon aims to understand the molecular mechanisms of LSD and develop new therapeutic approaches. Dr. Heldermon coordinates the necessary multi-disciplinary and personalized therapy approaches for affected patients and their families. Dr. Heldermon also collaborates with LSD centers throughout the United States and around the world to bring advances from basic research to clinical care.
What are lysosomal storage diseases?
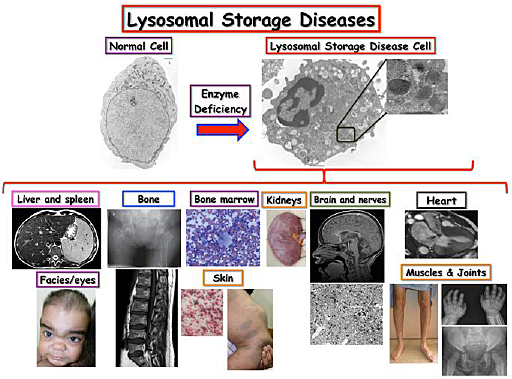
Lysosomal storage diseases are caused by the malfunction enzymes that degrade several substances inhuman cells. These enzymes are found in sac-like structures inside cells called lysosomes. Lysosomes function as recycling units of each cell, which contain hundreds of thousands of them. Lysosomes harbor specific enzymes that breakdown several substances, including proteins, sugars, and lipids into simple products that the cell then utilizes to re-build these substances (Fig.1). Lysosomes and other related structures called endosomes are also essential for the transport or “trafficking” of different substances inside each cell. Each of lysosomal enzymes has specific substances that they are capable of degrading. In case there is a deficiency in one of these enzymes, there is a buildup of the substances, which these enzymes normally cleave, resulting initially in dysfunctional lysosomes, but systematically causing an aberrant function of an entire cell. Further, such defects in cells are then reflected in the malfunction of organs, which consist of these cells, resulting in severe and progressive health problems.
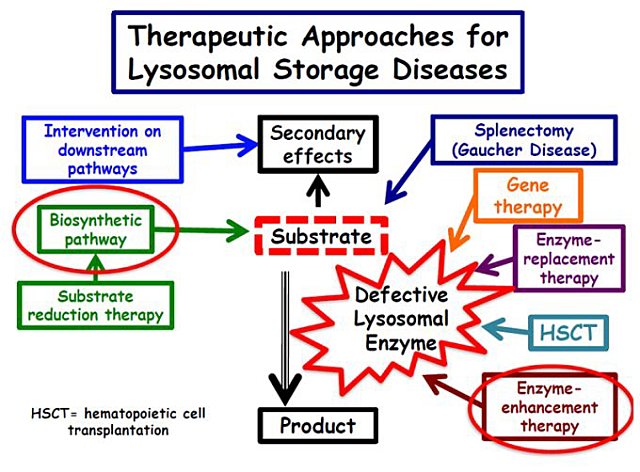
In general, the supportive/symptomatic treatment deals with the secondary effects of lysosomal enzyme deficiency. The specific treatments address either the accumulated substance (surgical procedures and substrate reduction therapy, SRT, a defective lysosomal enzyme by gene therapy (replacing the altered gene that generates a defective enzyme) or by enzyme augmentation therapy. This therapy is based on providing the normal enzyme through enzyme replacement therapy (ERT) or hematopoietic stem cell that provides donor cells that produce the normal that is taken by the patient’s disease cells with enlarged and dysfunctional lysosomes. A novel type of treatment is focused on enhancing the activity of the deficient enzyme that can still have a small but still insufficient enzyme activity.
Almost 60 LSD have been described as known lysosomal storage diseases. Some common LSD include:
Fabry disease
Results from the accumulation of globotriaosylceramide. It is known as X-linked genetic disease, affects both male and females, causing pain, gastrointestinal problems, progressive kidney, heart and pulmonary problems, chronic pain and is associated with characteristic dark red skin spots
Gaucher disease
Progressive LSD causing enlargement of spleen and liver, as well as bone lesions. Some forms of Gaucher disease also affect the brain, leading to severe neurological conditions
Neuronal ceroid lipofuscinosis (also known as Batten’s disease)
This is a group of neurodegenerative disorders named for the way the brain cells of patients look under the microscope. They mostly present in the first years of life and are characterized by visual failure, seizures and progressive cognitive decline. There is now a treatment for one subtype.
Niemann-Pick C disease
Results in progressive neurological condition along with lung disease, as well as enlargement of the spleen and liver
Pompe disease
Frequently fatal condition, which is presented in infancy. It results from glycogen build up in the heart and other organs, initially also known as acid maltase deficiency. If it manifests in childhood and adulthood, Pompe can cause progressive weakness of shoulder, hips, and respiratory muscles
Metachromatic leukodystrophy and Krabbe disease
Devastating LSD that results in progressive and neurodegenerative conditions. Recently adults have been described with milder forms that present with neuropathies and psychiatric problems.
The LSDs are relatively rare genetic disorders affecting 1 in 2,000-3,000 live births. Some specific LSD can occur more often in certain ethnic groups including Ashkenazi Jew (Gaucher, Niemann-Pick A, mucolipidosis-IV, Tay-Sachs), French-Canadian (Tay-Sachs), Cajun (Tay-Sachs), infantile neuronal lipofuscinosis (Finland). Because these diseases follow several patterns of inheritance, a person’s risk of passing this condition on to his or her children depends on the disease and the individual’s family background.
LSD treatment
Are there treatments available for patients with LSDs?
The treatment of LSDs can be divided into supportive/symptomatic and specific treatment (Fig.2).
Both treatment modalities complement each other, and together they aim the best quality-of-life for the affected patients and their families.
Supportive treatment involves the multi-disciplinary care of different specialties. Usually, the multiple problems that LSD patients develop require symptomatic/supportive treatment to address the secondary effects of the accumulation of a substance in lysosomes. Several specialties have an essential role in determining the clinical outcome and quality of life for patients with LSDs, including cardiology, anesthesia, orthopedics, otorhinolaryngology, ophthalmology, neurology, neurosurgery, as well as paramedical groups including physiotherapy, occupational therapy, nutrition, audiology, speech therapy, and psychology.
Lysosomal storage diseases (LSDs) are genetic diseases caused by specific enzyme deficiencies that result in the buildup of undegraded substance inside cell organelles called lysosomes, which are sac-like structures present in every cell in our body. Lysosomes are recycling units, i.e., they are responsible for breaking down lipids, sugars, and proteins into small units that can be re-utilized by the cell. Lysosomes and related endosomes are responsible for intracellular trafficking that is essential for cell function. The figure shows a blood cell from a healthy individual and a patient with an LSD, showing the enlargement of several lysosomes containing the undegraded substance. Enlargement of lysosomes ultimately results in the impairment of lysosomal function and consequently cellular function in multiple organs and systems. Therefore, LSDs manifest as systemic diseases in patients with multiple and progressive neurological, renal, cardiovascular, gastrointestinal, musculoskeletal, ophthalmological, cutaneous and respiratory problems.
Enzyme-augmentation therapy
Hematopoietic stem cell transplantation (HSCT)
The aim is to use donor-derived cells as a source of enzyme.
The cell source can come from bone marrow, or umbilical cord donor macrophages can infiltrate diverse patients’ tissues including the central nervous system. These donor-cells secrete a normal lysosomal enzyme (able to break down the accumulated substrate) which is taken up by patients cells, reaching their enlarged lysosomes directly and cleaning them as the defect.
When done before a patient with severe form of disease starts to develop neuroregression, HSCThas shown to be efficacious in one specific LSDs, MPS type 1 and a treatment algorithm is described. However, it has not been proven to have the same result in most LSDs. Also, most data available is anecdotal or is based. In Krabbe disease and metachromatic leukodystrophy, the initial benefit has demonstrated, preventing the fulminant course of these neurodegenerative diseases.
Enzyme-replacement therapy (ERT)
- Eight LSDs are treated with ERT – Gaucher disease, Fabry disease, mucopolysaccharidoses types I,II, IV and VI, lysosomal acid lipase deficiency
- ERT was initially developed for Gaucher disease type 1 (without neurological problems) using glucocerebrosidase purified from human placentas. The development of recombinant enzyme produced in cultured cells increased the scale of the production. The primary limitation of ERT is its inability to treat neurological symptoms of most LSDs since the ERT agent is unable to cross the blood-brain barrier, which is a physiological barrier located in blood vessels located in the brain and spinal cord
- Patients on ERT require administration of the recombinant enzyme diluted in normal saline intravenously every one or two weeks.
- Evaluating different clinical outcomes for 6-12 months can note the response to ERT.
Substrate reduction therapy (SRT)
- SRT is based on reducing the production of the accumulating substance of a deficient lysosomal enzyme in LSDs
- Miglustat and eliglustat are SRT FDA-approved agents to treat patients who are unable to tolerate ERT for Gaucher disease
- It reduces the production of several SRT agents
- In patients with Niemann–Pick type C disease, miglustat demonstrates stabilization of disease progression and is now licensed in Europe for the treatment of this disease
Enzyme enhancement therapy
- In most LSDs, the disease causes health problems only once the enzyme efficiency is below 10–15% of normal enzyme
- Enzyme-enhancement therapy is based on specific small molecules, which enhance the activity of the deficient enzyme when delivered to lysosomes. Increasing residual enzyme activity by only a few percentage may have profound clinical effects.
- The drugs are still in clinical trials, but they can be available for this type of treatment
What are the limitations of the current treatment for LSDs?
- The ERT is not able to treat neurological problems
- The decision to start ERT for Fabry disease, Gaucher disease (type I) and late-onset Pompe disease is based on patients symptoms or deterioration of those over time in each affected patient. The treatment decision is made in conjunction with patient and their family members
- Early initiation of treatment for mucopolysaccharidosis MPS-I, MPS-II and MPS-VI results in clinical benefit for patients with these conditions
- Although ERT is not effective for treating neurological problems, it may bring significant improvement in the quality of life of patients with severe clinical forms of MPS-II, MPS-II, MPS-III and neuronopathic Gaucher disease
- Pregnancy is not considered a contraindication for ERT in non-neuronopathic Gaucher disease type I and Fabry disease. At present, there are no reports evaluating the ERT during pregnancy in mucopolysaccharidosis MPS-I, MPS-II and MPS-VI
- For MPS-I, specific criteria for offering HSCT and ERT are well-established